Choroba Fabry’ego
Choroba Fabry’ego to lizosomalna choroba spichrzeniowa dziedziczona w sposób recesywny sprzężony z chromosomem X, spowodowana mutacją w genie GLA. W wyniku tej mutacji dochodzi do niedoboru enzymu alfa-galaktozydazy A, który odpowiada za rozkład tłuszczowych substancji. Niedobór enzymu prowadzi do nagromadzenia się globotriaozyloceramidu w różnych narządach.
Choroba Fabry’ego należy do grupy ultrarzadkich mukopolisacharydoz. Może powodować silne napady piekącego, przeszywającego bólu w dystalnych częściach kończyn, promieniujące do innych części ciała.
Charakterystycznymi objawami są również zaburzenia termoregulacji i pocenia się, nawet w sytuacjach stresowych. Pacjenci najczęściej umierają przedwcześnie z powodu uszkodzeń serca lub mózgu, zazwyczaj przed 50. rokiem życia. Ze względu na niespecyficzny charakter objawów, postawienie właściwej diagnozy może zająć wiele lat.
Przyczyny choroby Fabry’ego
Choroba Fabry’ego to rzadka lizosomalna choroba spichrzeniowa. Jej przyczyną jest mutacja w genie GLA, prowadząca do całkowitego braku lub znacznego niedoboru aktywności enzymu alfa-galaktozydazy A.
W efekcie dochodzi do odkładania się globotriaozyloceramidu (GL-3) w komórkach różnych tkanek i narządów, ponieważ brak enzymu uniemożliwia jego rozkład. Nagromadzenie GL-3 zaburza funkcjonowanie zajętych narządów i prowadzi do powikłań.
Choroba ta jest sprzężona z chromosomem X, co oznacza, że częściej występuje u mężczyzn. Jednak heterozygotyczne kobiety również mogą wykazywać objawy choroby, choć ich nasilenie bywa zróżnicowane. U kobiet objawy kliniczne zazwyczaj pojawiają się później niż u mężczyzn.
Diagnostyka choroby Fabry’ego
Aby potwierdzić diagnozę choroby Fabry’ego u mężczyzn, mierzy się aktywność enzymu alfa-galaktozydazy A w osoczu, leukocytach lub fibroblastach. W przypadku wykrycia niedoboru enzymu, przeprowadza się badanie genetyczne. U kobiet, ponieważ aktywność alfa-galaktozydazy A może mieścić się w zakresie normy, konieczne jest badanie genetyczne w celu identyfikacji nosicielek wadliwego genu odpowiedzialnego za chorobę Fabry’ego.
Objawy choroby Fabry’ego
Choroba występuje w dwóch fenotypach:
- Fenotyp klasyczny – Częstszy u mężczyzn, charakteryzuje się brakiem lub minimalną aktywnością enzymu, a objawy pojawiają się już w dzieciństwie lub okresie dojrzewania.
- Fenotyp nietypowy (non-classic) – Objawy pojawiają się później w życiu z powodu mniejszego niedoboru enzymu alfa-galaktozydazy A.
Objawy występujące w okresie dojrzewania to m.in.:
- Nietolerancja wysokich temperatur
- Zaburzenia pocenia się
- Hipertermia
- Parestezje (silny, piekący ból w dystalnych częściach kończyn)
- Bóle brzucha, zaparcia, biegunki, nudności i wymioty
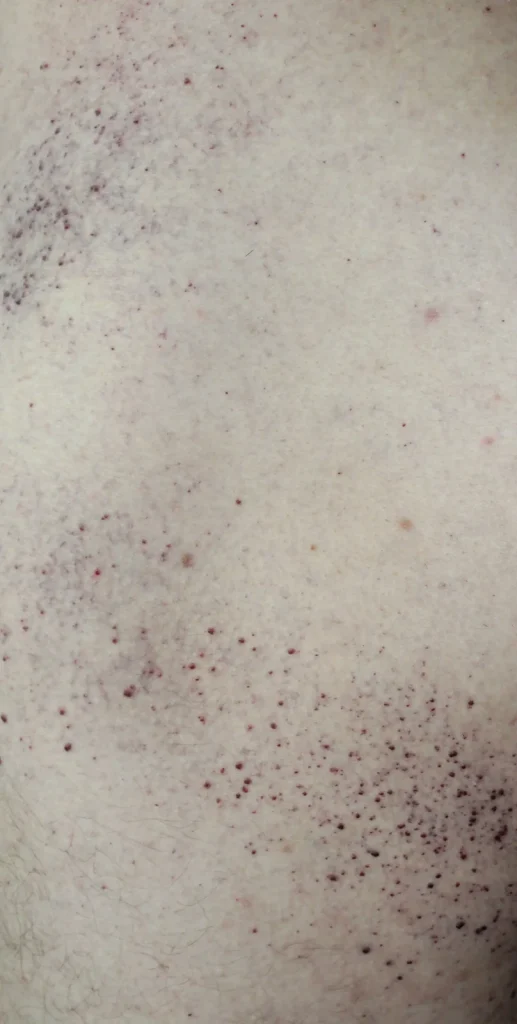
- Zmiany skórne w okolicy ud, pośladków i podbrzusza w postaci czerwono-fioletowych wykwitów (angiokeratoma)
- Zmętnienie rogówki, zaćma oraz postępująca utrata słuchu
W wieku dorosłym objawy obejmują:
- Strukturalne i czynnościowe nieprawidłowości serca (przerost lewej komory, arytmie, zaburzenia przewodzenia)
- Uszkodzenia nerek, początkowo objawiające się białkomoczem i mikroalbuminurią
- Przewlekłe zmęczenie, szumy uszne i zawroty głowy
Dodatkowo często występują zmiany w zachowaniu, takie jak lęk, izolacja społeczna, depresja i poczucie winy. Wiele z tych objawów pojawia się jeszcze przed postawieniem diagnozy, ponieważ pacjenci z chorobą Fabry’ego – podobnie jak osoby z innymi chorobami rzadkimi – często doświadczają wieloletnich opóźnień diagnostycznych, co wpływa zarówno na ich zdrowie fizyczne, jak i psychiczne. Badania pokazują, że jakość życia pacjentów z chorobą Fabry’ego jest istotnie niższa w porównaniu do populacji ogólnej
Rokowanie
Najczęstszymi przyczynami zgonu u pacjentów z chorobą Fabry’ego są udar mózgu i zawał serca. W porównaniu do ogólnej populacji, mężczyźni z chorobą Fabry’ego żyją średnio o 15–20 lat krócej, natomiast kobiety o około 6–10 lat krócej.
Opcje leczenia
Standardowym leczeniem choroby Fabry’ego jest enzymatyczna terapia zastępcza (ERT) z wykorzystaniem agalsidazy alfa lub beta. Ze względu na wielonarządowy charakter choroby, konieczne jest również stosowanie terapii wspomagających, które pozwalają kontrolować powikłania i zapobiegać dalszemu pogorszeniu stanu zdrowia.
Terapia ERT może spowolnić postęp choroby i umożliwić pacjentom utrzymanie aktywności rodzinnej, zawodowej i społecznej.
Dodatkowo, migalastat został zatwierdzony do leczenia choroby Fabry’ego. W przeciwieństwie do dożylnej ERT, migalastat jest lekiem doustnym. Jednak może być stosowany tylko u pacjentów z określonymi mutacjami, które są wrażliwe na ten lek – dotyczy to około 35–50% chorych.
Terapie wspomagające w chorobie Fabry’ego obejmują:
- Leczenie bólu, głównie farmakologiczne
- Neuroprotekcję (inhibitory ACE oraz blokery receptora angiotensyny)
- Leki antyarytmiczne
- Wszczepienie rozrusznika lub kardiowertera-defibrylatora (ICD) w przypadku ciężkich zaburzeń sercowo-naczyniowych
- Dializoterapię lub przeszczep nerki przy zaawansowanej niewydolności nerek
Leczenie choroby Fabry’ego jest prowadzone przez całe życie, a wczesna interwencja znacząco zwiększa szanse na opóźnienie lub uniknięcie poważnych powikłań.
Występowanie
Na świecie częstość występowania choroby Fabry’ego szacuje się na 1 na 40 000 do 1 na 170 000 osób. Jednak z powodu złożoności objawów oraz trudności diagnostycznych, dane te mogą być niedoszacowane.